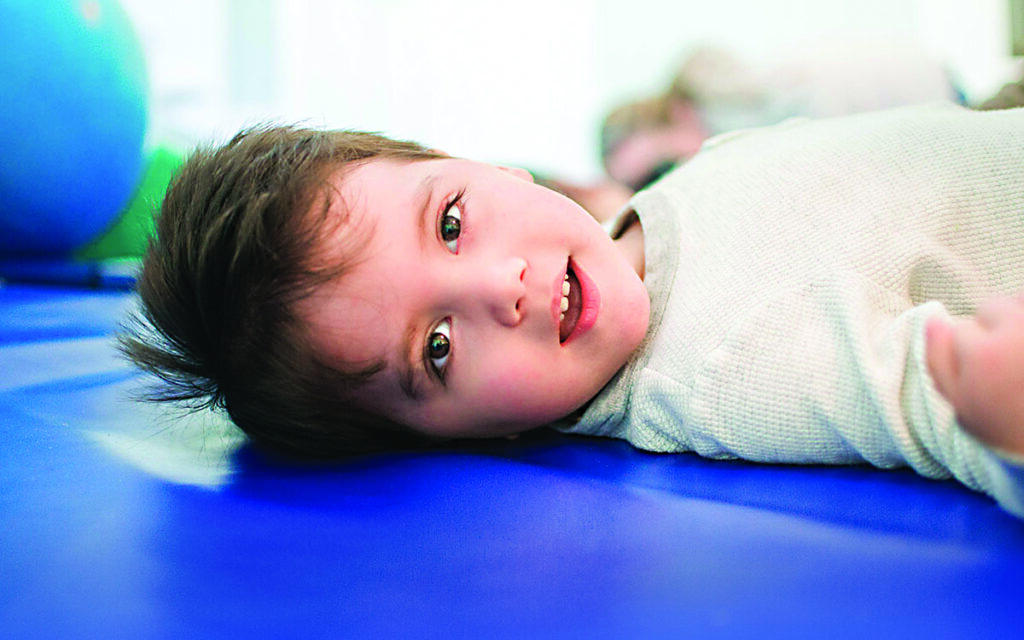
Ci sono malattie così rare che non hanno ancora avuto un nome tutto loro. È il caso di una patologia che non solo è rarissima ma è anche poco nota, al punto che viene ancora identificata con la sua causa scatenante: la mutazione del gene GNAO1. Al momento ci sono meno di duecento malati in tutti il mondo. L’esordio pediatrico è precoce e i sintomi iniziali vanno dall’epilessia ai disturbi del movimento, e possono comparire già nei primi giorni di vita. Nel 2013 un gruppo di ricerca giapponese è riuscito a identificare una mutazione casuale a carico di uno specifico gene, che si chiama GNAO1. Questo acronimo sta per G Protein Subunit Alpha O1 perché il gene codifica per la subunità Alpha O1 della proteina G. Una proteina fondamentale coinvolta nella complessa catena di comunicazione molecolare che avviene tra le cellule, in particolar modo nella trasmissione del segnale nelle cellule nervose. Tempo addietro questa patologia era considerata una grave encefalopatia epilettica ma negli anni è stata riscontrata anche a forme più lievi di epilessia, o per niente associata a epilessia. Altri sintomi molto comuni sono un ritardo psicomotorio, ipotonia e movimenti involontari rapidi, caotici, di tipo coreico, che durano anche alcune ore e lasciano i piccoli pazienti fortemente disabilitanti. Non esiste una unica mutazione del gene GNAO1, e a essere coinvolte sono zone diverse del gene. Le diverse tipologie e intensità dei sintomi che si osservano nei bambini affetti da GNAO1 potrebbero essere correlate proprio al tipo di mutazione. Il difficile inquadramento clinico legato alla grande variabilità clinica riportata in letteratura, rappresenta ancora l’aspetto più complesso perché non si riesce capire come mai i sintomi siano così diversi da bambino a bambino, e perché differiscano anche all’interno della stessa variante della mutazione. Oggi si sa che un difetto a carico della subunità della proteina G provoca l’attenuazione della trasmissione del segnale fra le cellule nervose, e ciò è probabilmente alla base della comparsa delle crisi epilettiche, mentre un altro difetto capace di amplificare il segnale induce i movimenti incontrollati e accelerati di tipo coreico. In realtà, nonostante la crescita dei gruppi di ricerca e di clinici che stanno indagando su questa nuova patologia, si sa ancora troppo poco sulla GNAO1 e della sua epidemiologia, in particolare non si è certi del quadro clinico generale e del suo decorso. Ciò fa pensare che il trasferimento delle conoscenze dalla ricerca di base allo sviluppo clinico di terapie efficaci prenderà ancora molto tempo. Nel frattempo numerosi studiosi pensano che non si tratti di una malattia degenerativa, ma di una condizione stabile nel tempo, che è causa di una grave disabilità. Nel 2016, negli Stati Uniti è stata fondata dai genitori di bambini GNAO1 un’associazione internazionale, la Bow Foundation, che si è posta come missione la creazione di un network di famiglie, ricercatori e clinici, per finanziare progetti di ricerca e richiamare l’attenzione generale sulla malattia. In Italia a occuparsi della GNAO1 ci sono la Fondazione Telethon, l’Osservatorio Malattie Rare, la Fondazione Policlinico Gemelli di Roma, dall’Azienda Ospedaliero-Universitaria Meyer di Firenze, l’Ospedale Pediatrico bambino Gesù e l’Azienda Ospedaliero – Universitaria Policlinico Umberto I di Roma, che operano in collaborazione con l’Associazione Famiglie GNAO1 APS che ha organizzato in Italia il primo Congresso Europeo con l’obiettivo primario di coinvolgere le comunità di medici, ricercatori ed esperti di riabilitazione in Italia, ed in Europa, per gettare le fondamenta di un polo avanzato e di eccellenza per la ricerca sulla GNAO1. Tra i relatori neurologi, genetisti, ricercatori e terapisti provenienti dall’Italia, Olanda, Germania, Svizzera e Inghilterra. Divulgare la conoscenza di questa rara patologia, può servire a migliorare l’assistenza e contribuire ad accelerare il percorso della ricerca.
In realtà, nonostante la crescita dei gruppi di ricerca e di clinici che stanno indagando su questa nuova patologia, si sa ancora troppo poco sulla GNAO1 e della sua epidemiologia, in particolare non si è certi del quadro clinico generale e del suo decorso. Ciò fa pensare che il trasferimento delle conoscenze dalla ricerca di base allo sviluppo clinico di terapie efficaci prenderà ancora molto tempo. Nel frattempo numerosi studiosi pensano che non si tratti di una malattia degenerativa, ma di una condizione stabile nel tempo, che è causa di una grave disabilità. Nel 2016, negli Stati Uniti è stata fondata dai genitori di bambini GNAO1 un’associazione internazionale, la Bow Foundation, che si è posta come missione la creazione di un network di famiglie, ricercatori e clinici, per finanziare progetti di ricerca e richiamare l’attenzione generale sulla malattia. In Italia a occuparsi della GNAO1 ci sono la Fondazione Telethon, l’Osservatorio Malattie Rare, la Fondazione Policlinico Gemelli di Roma, dall’Azienda Ospedaliero-Universitaria Meyer di Firenze, l’Ospedale Pediatrico bambino Gesù e l’Azienda Ospedaliero – Universitaria Policlinico Umberto I di Roma, che operano in collaborazione con l’Associazione Famiglie GNAO1 APS che ha organizzato in Italia il primo Congresso Europeo con l’obiettivo primario di coinvolgere le comunità di medici, ricercatori ed esperti di riabilitazione in Italia, ed in Europa, per gettare le fondamenta di un polo avanzato e di eccellenza per la ricerca sulla GNAO1. Tra i relatori neurologi, genetisti, ricercatori e terapisti provenienti dall’Italia, Olanda, Germania, Svizzera e Inghilterra. Divulgare la conoscenza di questa rara patologia, può servire a migliorare l’assistenza e contribuire ad accelerare il percorso della ricerca.
08
Mar
Mar